
In the field of fragment-based drug discovery (FBDD), biophysical methods such as NMR, native MS, high-concentration biochemical screens (HCS), thermal shift assays (TSA), or surface plasmon resonance (SPR) are usually used as pre-screening techniques to screen fragment libraries to identify the most promising binders.
However, employing a subsequent cascade of these methods prior to X-ray crystallography has been proven rather misleading as they might miss an important fraction of binding fragments that could be observed in crystal structures. In addition, the overlapping hits resulting from biophysical methods compared to X-ray crystallography have been rather poor.
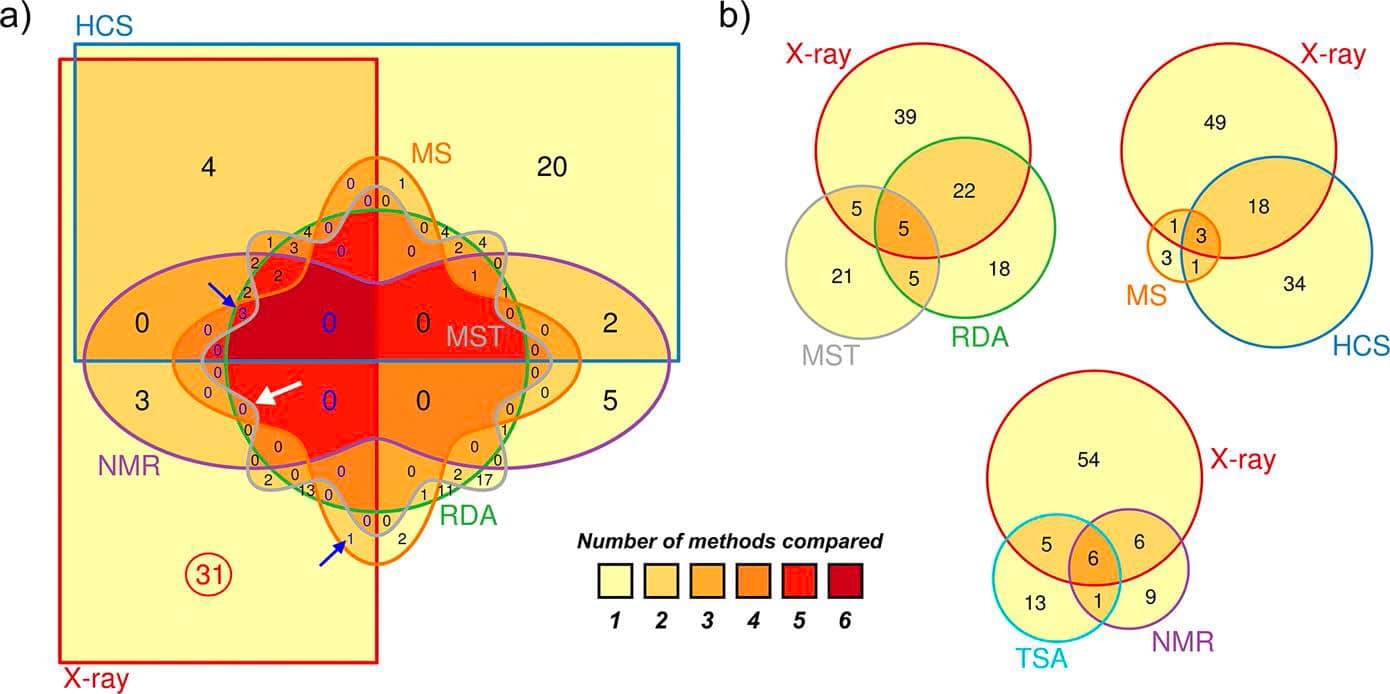
Venn diagrams show low overlaps between hits identified in X-ray crystallography and other biophysical methods.
Among all possible screening methods, X-ray crystallography is not only the most sensitive method for detection of fragment is binding. It also reveals the geometry of binding as precise three-dimensional positions of atoms in a protein structure. This is essential to investigate the accessible chemical space in a protein-ligand complex for further development of the initial hit into a lead candidate. Highly resolved crystal structures can also reveal the exact position of water molecules and water networks in the binding site. In this regard, it has been shown that water molecules in fixed position and their displacement can be an important data in structure-based lead discovery.
Remarkably, X-ray protein crystallography is not only able to detect high- but also low-affinity binders that cannot be detected by any other method.
Only after having precise structural information about the initial hit and exploring the binding pocket, other biophysical methods can be used to further characterize the protein-ligand complex and improve affinity, potency, and binding kinetics of fragment expansion campaigns.

Osborne, J.; Jhoti, H. et al., 2020
Thanks to third-generation synchrotron sources and the latest methodological improvements in automated crystal mounting systems, data collection, and processing, it has become increasingly feasible to screen entire libraries of fragments efficiently. Although automation enables efficient data collection, the most important part before data collection is the development of a high-quality crystallization and high-performance soaking systems. This could be compared to assay development for high-throughput screening campaigns. The quality of crystals in terms of diffraction and reproducibility has to deliver consistent and comparable results. It’s not about getting one crystal at sufficient quality, but rather hundreds of crystals, which enables a screening at all. This requires specific expertise, technology and a broad range of experience.
With novel workflows and pipelines, like FastForward, it is possible to utilize the large amount of collected data in a smart way. In fact, starting from the raw data given by X-ray protein crystallography, it is possible to process and refine the datasets of fragment library screens toward high-quality 3D models to design higher–affinity compounds in a short period of time.