Magnet for the Needle in Haystack: “Crystal Structure First” Fragment Hits Unlock Active Chemical Matter Using Targeted Exploration of Vast Chemical Spaces
We introduce a novel, multidisciplinary method to identify active molecules from purchasable chemical space.Four protein kinase A small-molecule fragment complexes were used as the starting point for a template-based docking screen utilizing multibillion REAL Space. In just 9 weeks, our unique fragment-to-hit technique had an overall success rate of 40%, with the most active follow-up showing a 13,500-fold increase in affinity.
IMPROVING THE SELECTIVITY OF 3-AMIDINOPHENYLALANINE-DERIVED MATRIPTASE INHIBITORS
Novel 3-amidinophenylalanine-derived matriptase inhibitors with enhanced selectivity against thrombin and factor Xa were created using a rational structure-based methodology. Three crystal structures of related substances in trypsin and thrombin as well as the crystal structure of a selective monobasic matriptase inhibitor in association with matriptase have been identified.
Conceptional Design of Self-Assembling Bisubstrate-like Inhibitorsof Protein Kinase A Resulting in a Boronic Acid Glutamate Linkage
A potential method for producing self-assembled bisubstrate-like inhibitors in the binding pocket of cAMP-dependent protein kinase is the spontaneous esterification of boronic acids with polyols. Multiple crystal structures of the protein, covalently modified with residues such as a ribopyranosylated serine and threonine or a phenylboronic acid attached to lysine via amide bonds, were determined with the phenylboronic acid-linked Fasudil.
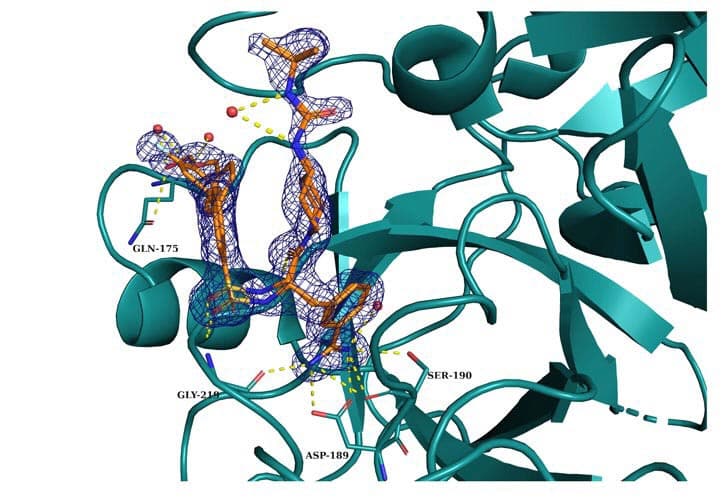
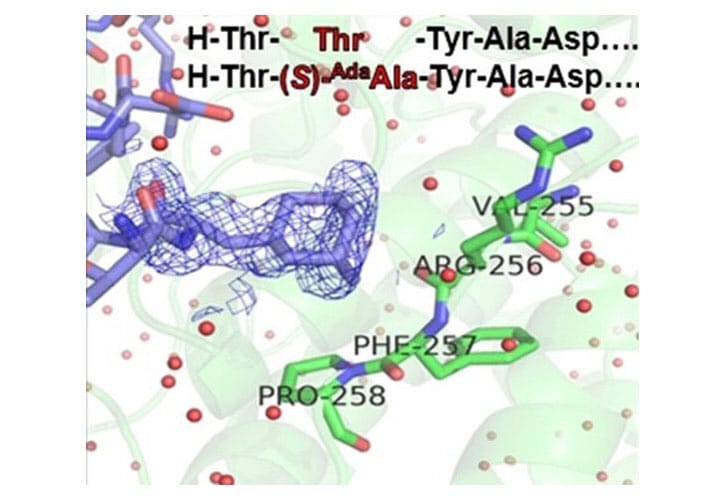
DiamondoidAminoAcid-Based Peptide Kinase A Inhibitor Analogues
By altering their secondary structure and offering a distinctive spatial arrangement of exit vectors to attach other moieties, diamondoid amino acids are incorporated into peptide-like medications as a general technique to increase their lipophilicity, membrane permeability, and metabolic stability. We created five brand-new peptidic protein kinase A inhibitors and put them through binding tests, NMR spectroscopy, and crystal structure analysis to learn more about their characteristics.
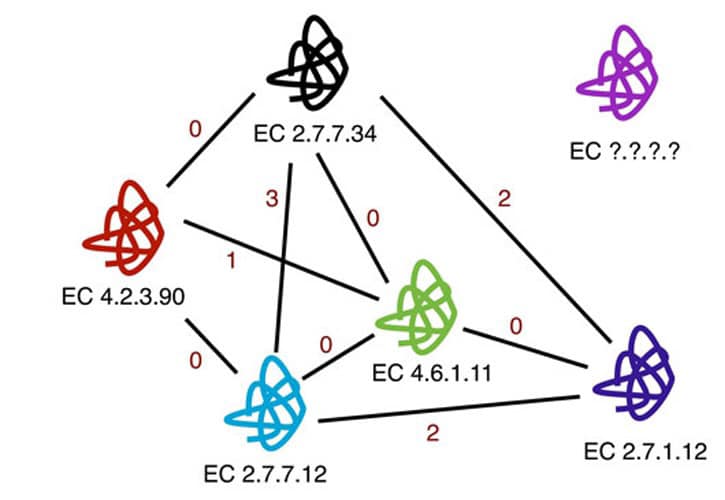
Identification of functionally related enzymes by learning-to-rank methods
This work demonstrates how the use of kernel-based learning algorithms can significantly enhance rankings of biological function. The method demonstrates that it is possible to identify statistical correlations between an enzyme’s active cleft and its biological function. A series of similarity measurements that take advantage of knowledge about cavities on the surface of enzymes show a constant and considerable improvement, according to the findings of a sizable number of studies.
Cavities tell more than sequences: exploring functional relationships of proteases via binding pockets
Proteins can be categorized according to the characteristics of their binding sites, which is a viable method for gathering data for selectivity modeling. In order to achieve this goal, the workflow clusterScore has been created. It explores key clustering technique factors and enables precise protein categorization utilizing protein pocket information. These findings highlight the significance of binding-site data, which should be taken into account when designing ligands throughout lead optimization cycles.
Investigation of specificity determinants in bacterial tRNA-guanine transglycosylase
The exchange of the genetically encoded guanine at the wobble position of the tRNAsHis, Tyr, Asp, Asn by the premodified base preQ1 is catalyzed by the bacterial enzyme tRNA-guanine transglycosylase (Tgt). The work shows proof that the Cys158Val mutation decreases the binding to preQ1 while keeping the affinity to guanine unaltered using enzyme kinetics and X-ray crystallography.
Novel type II fatty acid biosynthesis (FAS II) inhibitors as multistage antimalarial agents
Enoyl acyl carrier protein reductase of P. falciparum is a crucial enzyme for plasmodial type II fatty acid biosynthesis. We discovered a structurally unique family of FAS inhibitors via virtual screening. These two most promising derivatives were discovered to have IC50 values of 1.7 and 3.0 µM that limit the growth of blood-stage parasites and cause a more pronounced developmental retardation of liver-stage parasites than the gold-standard medication, primaquine.
The use of isothermal titration calorimetry & molecular dynamics to show variability in DNA transfection performance
This study has characterized the electrostatically driven self-assembly process of linear polymethacrylate polymers with DNA-generating nanocarriers for efficient gene transfection. A polymer’s effectiveness as a nanocarrier can be predicted from the flexibility of its structure using the combination of isothermal titration calorimetry and computational molecular dynamics, which has proven to be a useful method for carrier-payload interaction investigations.
Superposition and alignment of labeled point clouds
A labeled point cloud is a finite collection of points where each point has a discrete class label that denotes a particular property in addition to being connected to a position in three dimensions. We discuss the problem of similarity comparison between two labeled point clouds. As a result, we create a technique in this study for calculating pairwise or multiple alignments of labeled point clouds. The structural study of protein binding sites is done using the technique.